

A Substack on Sanger, Next Generation and Oxford Nanopore sequencing techniques
For understanding current and upcoming news

For anyone reading or simply following Kevin McKernan’s vital work on the subject matter of ‘what’s in the COVID modified mRNA product vials’, you will have come across various sequencing techniques and names like Illumina (Next Generation Sequencing), Sanger and Oxford Nanopore. I decided it’s important for you all who don’t have a background in genomics (like me!) to know a little bit more about what these sequencing methods involve, why they are different and how they are different. I think this is really important because of what’s coming in the future and the best weapon against totalitarianism is knowledge.
I will start with Sanger sequencing and Next Generation Sequencing. This is by no means going to be a comprehensive review; just a gander.
First of all, sequencing itself refers to determination of the sequence of bases, A, T, G and C in DNA in something. That something could be anything that is made up of bases, including us! We are not much more than a linear sequence of bases arranged in a very special ways. How weird is that?
As per technological techniques to accomplish the task of determining these sequences, Sanger came first. In 1977, thanks to a guy named Frederick Sanger, the evolution of sequencing began. Sanger sequencing is considered the gold standard of sequencing and is 99.9% accurate in calling bases, and it’s still pretty cost-effective for low sample numbers. It was pretty slow and tedious in the beginning, and required radioactive tags as per base, running gels, and preparing and reading x-ray film: all manual. You could sequence about 200 nucleotides in about 4 days from a very small sample size and only one DNA strand could be sequenced at a time.
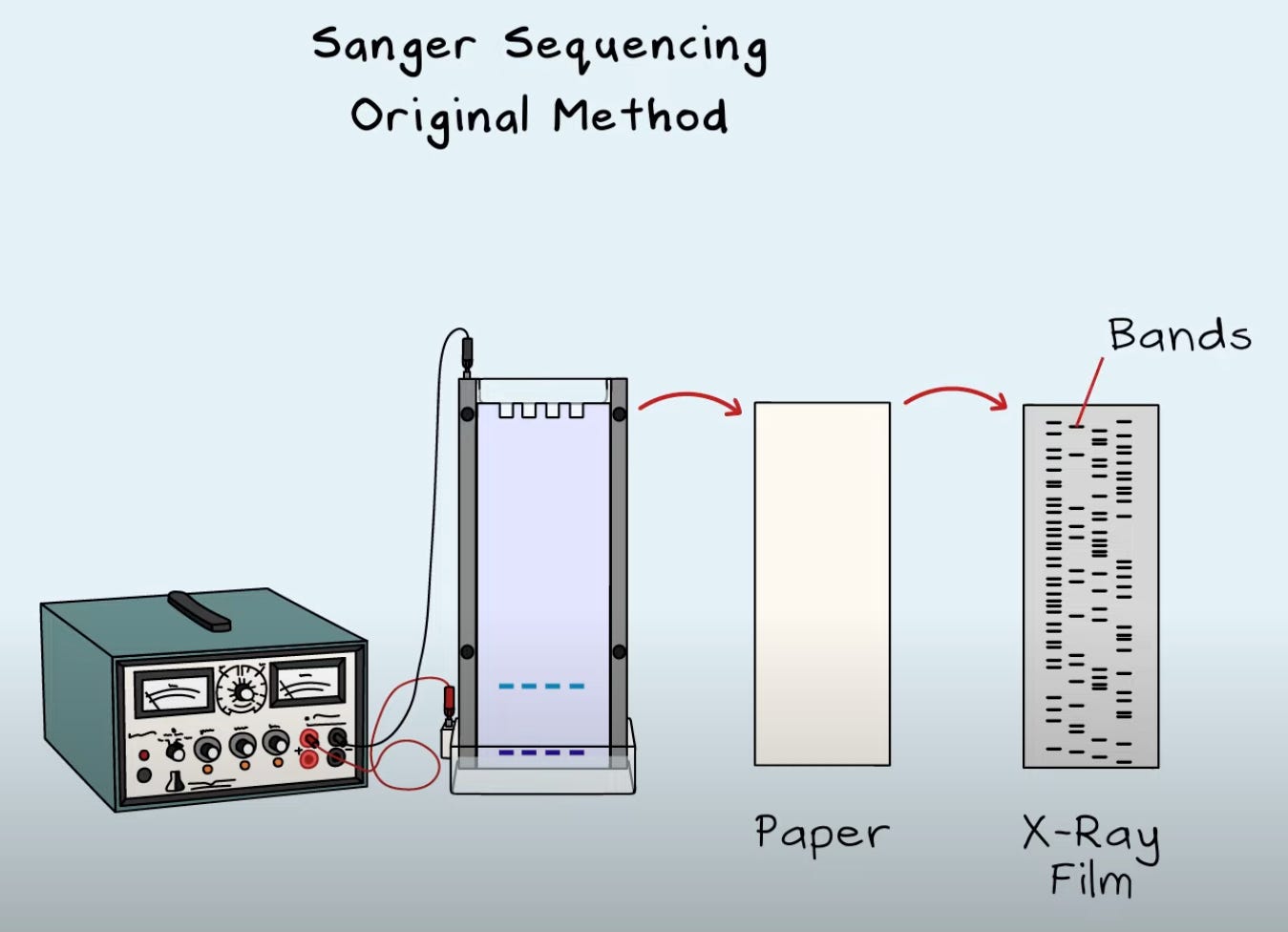
Scientific progress is amazing, and soon enough, most of the manual protocol became automated. The power of machines. In 1987, Applied Biosystems brought in fluorescent dyes as a safer alternative to radioactive tags. They built the AB370A machine that made sequencing faster through automation. Fluorescent dyes also meant you could use a different coloured dye as per A, T, G, C base making it possible to do stuff in one tube (Big Dye Terminators that came around in 1997), and increase the speed of the protocol even more. This method brought us up to about 450 base pair (bp) reads. Getting longer! I left out some things about heat-loving Taq polymerase (in addition to DNA polymerase) for building DNA, and about capillary electrophoresis (Beckman Coulter - 1989), but never mind that for now. It was in 1989 that Taq polymerase was introduced in the context of Sanger sequencing.
The evolution and optimization of various elements of Sanger sequencing eventually translated into needing less sample for your reaction, having faster runs, automation of sample loading, testing more samples at once all, etc… all to end up with even more bases being read: up to 600 bp now!

Now it’s 1990, and it’s time to talk about the Human Genome Project (HGP). At this time, less than 2% of the human genome had been sequenced. The Human Genome Project was part of no small feat that resulted in the eventual sequencing of all 3.2 billion bases of the human genome. Imagine doing that at 600 bp sequenced per experiment! By 1998, about 6% of the human genome had been sequenced, nice and slow. At about the same time, Applied Biosystems partnered with Craig Venter’s non-profit company: The Institute of Genomic Research (TIGR) in 1998, to form Celera. They bought 230 Applied Biosystems ABI Prism 3700 machines with the aim to sequence the human genome faster than the dudes involved in the HGP. Don’t ask me to explain who had what role with regard to ethics, or who worked with whom in this endeavor, because I don’t know the details. “The political, personal, and ethical conflicts of the race between the public and private sectors have been notable.”1 I am sure it was. Collins’ role, also noted.
Anyhoo, with all this competition and Celera selling sequence data to make money to keep the work going, the human genome was finally sequenced. Each run of 96 samples in the ABI Prism 3700 machine took less than 2.5 hours and yielded 800 bp of sequence for each sample. 1,536 samples could be sequenced daily. So by 2001, both Celera and the HGP had published their results. Now that’s progress.
See what competition can do? Imagine a world where there’s only the NIH in control of everything. We need independent companies, and we need competition.
With the human genome in hand, we had a reference sequence. This is what made further evolution possible.
Next Generation Sequencing (NGS) came … next. It is deep, massively parallel dude. It’s cool because it’s fast. It involves amplification of short DNA fragments ultimately aligned against a reference genome. This reference genome is the result of the work from the Human Genome Project/Celera which ironically, was created using (modified) Sanger Sequencing!
Instead of the decades that it took to finally sequence the human genome, in a single day using NGS, we can sequence an entire human genome. In fact, we could technically sequence 128 of them. The reason it is so much faster now is because NGS allows for millions of DNA fragments or strands to be sequenced simultaneously, instead of doing one at a time as with the original Sanger sequencing. You can do billions of reads per sample with NGS with 16 terabytes of read coverage, as opposed to 1 read per sample of only 300-850 base coverage with Sanger!
To begin, a library is created as a composite of shorter DNA fragments that are sourced from a long stretch of DNA. The long DNA is cut into shorter segments of specific lengths using sound waves or enzymes, and the shorter sequences are capped with adapters complete with identifiers (the index).
Here’s a video that explains a little bit about it. It’s not necessary for you to know all of the details, but the important things to know is that NGS is super fast because there are millions, even billions of parallel sequencing reactions going on at once. It involves the reference DNA sequence, the creation of a library, a flow cell with bound oligonucleotides with complementary sequences to bind the adapters bookending the short DNA fragments, creation of multiple copies of each fragment (using PCR and bridge building (amplification of the fragments)), sequencing, and alignment to the reference genome.
So with NGS, we have gone from being able to sequences up to 200 bp, to 3.2 billion bp in a few decades and in a fraction of the time.
But it gets even better.
What might be even cooler still, is Oxford Nanopore sequencing. If/when I become a genomics person, I will use nothing but this method.
This method uses a protein nanopore that mimics the membrane proteins in cell membranes.2 A current is generated through the nanopore embedded in a well membrane (there are 2048 wells in any given flow cell) in an electrolyte solution, whereby the membrane has very high electrical resistance. By applying a potential across the membrane, a current can be generated through the nanopore, and thus single molecules that pass through the nanopore disrupt the current. The current can be measured and the molecule, identified. Basically what this means, is that you can measure the electrical disruption that a single nucleotide DNA induces as it passes through the pore.
These nanopores are arranged in arrays in a flow cell so that multiple nanopore experiments can be done simultaneously. It’s way faster than NGS because instead of 300 DNA read lengths at a time, it can do 100,000 at a time. At this rate of sequencing progress, all we’ll have to do is think what we want to sequence and poof! It’ll be done.
Watch this very cool animation with a cool soundtrack!
I hope this clears some things up with regard to sequencing techniques. It’s really just a taste of the tip of the iceberg, but I really just wanted to put some faces to to various sequencing techniques and establish a timeline of their evolution.
It’s so very exponential.
Maybe as an exercise, we can think of ways to make it even more efficient and perhaps, remote. I mean, we’ll always need a DNA sample to begin with, but imagine a time when you can simply know all your genes - including potential mutations - in an instant.
I find it fascinating, truly, but here I must reiterate and reinforce the idea that we absolutely need independent and competing true scientists in charge of this progress and the development of technology - especially technology that involves genes, bases, sequencing, and more. Monopoly entities have no incentive to share or to help others. They care not for progress - merely profit. We all know this.
Also, just to remind y’all, we’re facing the coming age of gene therapy that will be suggested as a way to swap out bad genes. Before they do this, they will need to sequence you to find out with genes need swappin’ out. How might they do that? Well, they might already have done so. This is speculation, but it did occur to me that that’s what those sharp DARPA brain stabbing-scwabby swabs might have been for. You remember, right? When everyone was going bat-pangolin-sheit crazy about ‘testing’ for a coronavirus. Makes you think, doesn’t it?
If you want to read a nice peer-reviewed article about the sequence of sequencing, you can find one here.
Peace out.
https://en.wikipedia.org/wiki/J._Craig_Venter_Institute
I used to study B-12 importers of E. coli bacteria as part of my second post doc. These machines are ATP-binding cassette (ABC) transporters and are embedded in cell membranes to enable import of vitamin B-12 into cells.
I will never forget when the sequencing and technique was poop pooed 💩, by an individual of stature importance and knowledge. Thanks Dr. Rose , you stood your ground …. Thank u for your knowledge and wisdom.
Thanks Dr Jessica Rose 🌹 it does explain a lot many I will view it many times over so I can understand it to explain to others around me but it also explains a lot about the health issues of the cOVID-19 injectables affecting the heart as you know electrical impulses are very critical to the function arrhythmia and a fib , the basic functionality of the heart ,
if that affects on a nano level it totally makes sense why there is such an increase (epidemic ) issues of heart elements in the world……