
Amplicon-to-amplicon variance in qPCR assays assessing DNA contamination in mRNA vaccines demonstrates over a 100-fold difference in quantitation
In other words, we reproduced the DNA impurity results again in a bunch of vials and some of DNA is at higher concentrations than others - important for detection methods ignored by regulators

I spent the past 2 days working very hard with Kevin McKernan and Charles Rixey at Medicinal Genomics. It was extremely fruitful and fun. Kevin is an excellent teacher - very patient and fun.
We tested 8 vials for DNA using 2 methods of quantification: qPCR and fluorometry and sequencing using Oxford Nanopore sequencing. We also did lots of controls and set up duplicates and triplicates. Many magnetic beads were used.
qPCR
qPCR, or quantitative PCR, is a laboratory technique that amplifies specific DNA sequences and measures their concentration in real-time during the amplification process, allowing for precise quantification of DNA copies by monitoring the fluorescence signal generated as the DNA is replicated. We used 4 different fluorophores. The fluorophores are attached to probes or intercalating dyes that bind to the DNA as it is amplified, and you then measure the fluorescence emitted by these fluorophores in real-time.
Fluorometry
Fluorometry is a method that quantifies DNA (or other biomolecules like RNA) by exciting them with light of a specific wavelength and measuring the emitted fluorescence, which is proportional to the concentration of the target molecule, providing a sensitive and specific measurement of DNA content. We used the intercalating dye PicoGreen (you can also use SYBR Green) to quantify the total dsDNA in our samples. This is an Invitrogen fluorometer like the one we used. It costs a bit but it’s so fast! And accurate.

Oxford nanopore sequencing
We used Oxford Nanopore sequencing to sequence the DNA. The idea is that individual molecules are passed through a tiny protein pore (nanopore) and changes in electrical current as the bases pass through are measured. This allows for real-time, long-read sequencing without the need for amplification or chemical labeling. We actually used a method that attaches a motor onto the lead sequence and fed in ATP to get the DNA through the pores. This helps to control the rate at which the DNA strand passes through the nanopore by processively unwinding the DNA and consuming ATP as an energy source.
So all in all, qPCR counts DNA copies as they’re made by watching glowies (different kind of glowies), fluorometry measures DNA (or RNA) by how much it glows when a dye sticks to it, and Oxford Nanopore sequencing reads DNA or RNA like a ribbon passing through a tiny hole, powered by a molecular motor.
All 3 methods involve a lot of preparation and cleaning of the DNA but the output results are amazingly fast for all 3. Like most things, preparation usually takes the longest. The thing that was most annoying and took the longest and that is in fact the most dangerous, was removing the metal rubber stop coverings from the unopened vials when we were prepping the vial contents.
Below is a screenshot of a schematic of a nanopore and how the DNA is fed through a single pore to read the nucleotides. We used the MinION MK1D sequencer. The computer we ran this on cost more than this device, for cost context.

So we have yet to compile our data but I am sure Kevin will have that done by the end of today. But for now, because everything takes place in real time, we know what we have. DNA aplenty in the vials we tested. The vials were from Japan. DNA should not be in the vials at those levels. Or at all since the COVID shots use the LNPs to get the genetic material into cells: a very efficient means to introduce genetic material indeed.
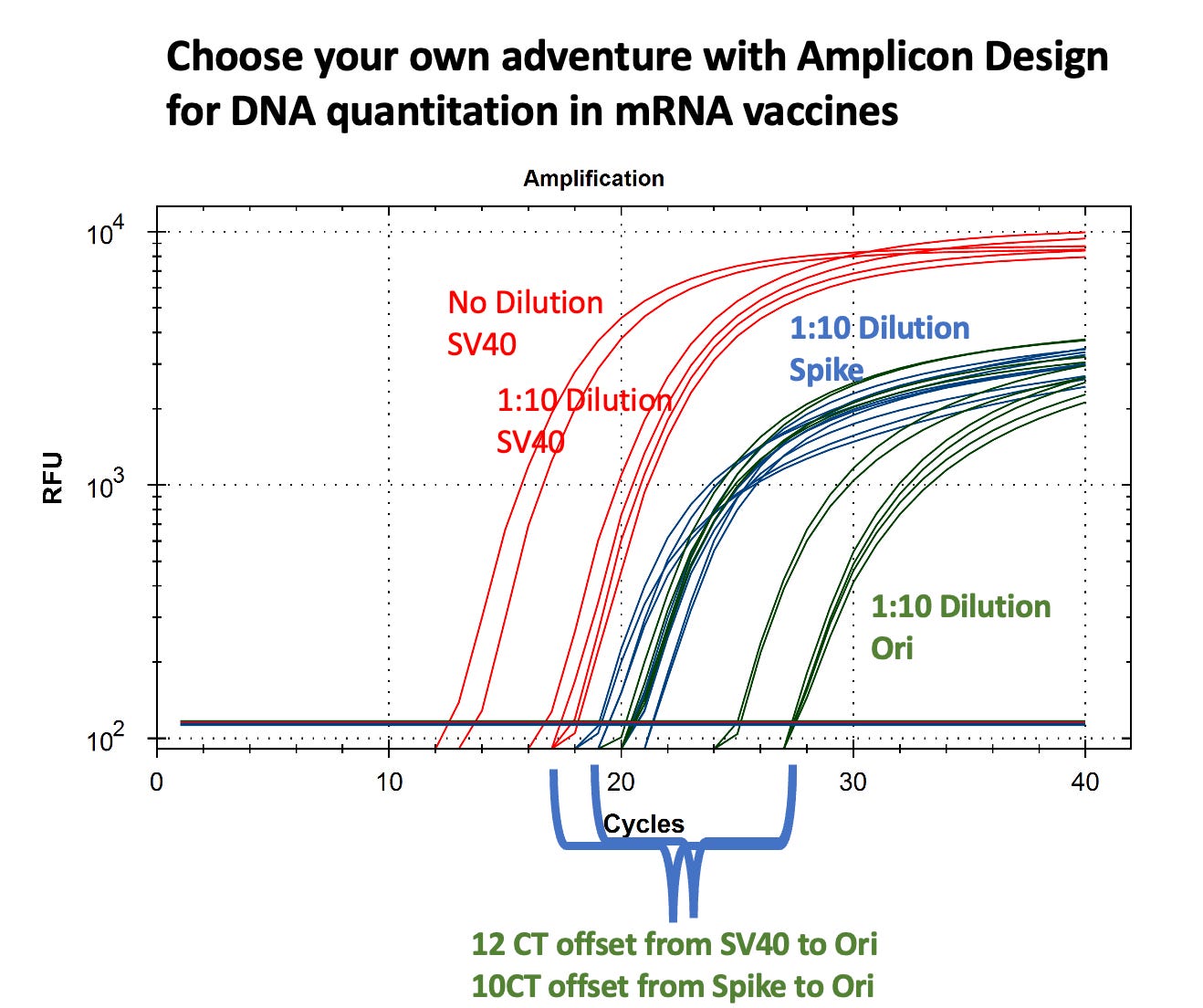
In the case of the qPCR as per the title of this article, we checked for amplification of Spike (blue), Ori (green) and SV40 (red) using specially-designed primers. Ori is the origin of replication in the plasmid that was used to make the N1-modRNA, Spike is spike, and SV40 is the SV40 promoter/enhancer region from the SV40 virus also in the plasmid (Spike/Ori are mostly RNA-derived; SV40 is plasmid-only). The qPCR results showed up to nine Cq (cycle threshold) differences between Spike-region amplicons and plasmid-vector (Ori) amplicons, corresponding to more than a 100-fold variation in quantitation. What this means is that there’s a lot more of one than the other, implying that there are big differences in DNase I digestion efficiency during the mN1-modRNA manufacturing process, particularly in the plasmid regions containing RNA:DNA hybrids.
Kevin has since re-done the assay using an RNase +/- study prior to qPCR. He did this to confirm that there wasn’t DNA being made by the RT polymerases during set-up since this can happen at high temperatures and it was quite warm in the lab, so it could have happened. We should have heat killed the RT polymerase mix before because the dwell time during the plate set up could have turned the RT enzyme on and made spike, but no plasmid which might have resulted in large offset in Spike to Ori that we saw. So today Kevin repeated the assay using RNase A treatment of the vaccines after TritonX lysis. But new results confirm that even this was not the only reason for the offsets we originally saw. So based on his results from today, the Spike Ct was a bit artificially low and that RT was on the go for Spike (and maybe Ori) making premature cDNA. There also appears to be lot-to-lot variation (differences in LNPs) so the uniform delay (even with variable deltas) confirms that the RT was actively producing DNA from vaccine RNA templates for both targets prior to the intentional heat-kill.
So he was able to rule out premature RT as the only explanation for the (early) huge offsets: there is real plasmid DNA in the sample He also strengthened our faith in our Ct differences and since RT can’t make SV40 from mRNA cuz there’s no SV40 in transcript, this confirms high-level pre-existing plasmid contamination, Spike/Ori are mostly RNA-derived; SV40 is plasmid-only.
This is the important part. Regulatory submissions reveal that residual DNA testing relies on a single qPCR target within the plasmid vector Kan region. This approach underestimates contamination levels because it does not assay the Spike region. Although EMA documentation discloses validation of a qPCR assay targeting the Spike insert, that assay is used only to confirm the presence of the insert, not to measure DNA content. This raises the question of why only single-amplicon quantitation is reported when multiple validated assays are available and produce significantly different results. It raises eyebrows, and fists. It’s impossible that this wasn’t done purposefully.
My post of our qPCR result video has gotten over 183,000 views in less than 24 hours. People want to know why the DNA issue wasn’t disclosed. They also want to know the implications in terms of clinical outcome. It’s cancer and inflammation, in a nutshell.

The video above shows qPCR DNA amplification results from Pfizer and Moderna COVID-19 vial samples revealing a high concentration of DNA fragments at a cycle threshold (Ct) of 9, which indicates a significant presence of contaminating DNA. We looked for Spike, Ori and SV40. There’s no SV40 in the Moderna samples.
The vials contain DNA fragments at levels far exceeding regulatory limits that imply potential risks associated with introducing foreign DNA fragments to cells via LNPs such as unintended immune responses or integration into the host genome to induce cancer pathways. SV40 also binds p53 and does other nasty stuff.
By the way, in addition to the fact that there shouldn’t be any DNA in these vials in the first place, there really shouldn’t be so much variation. Almost makes one wonder if they cherry-picked DNA quantitation methodologies to miss detecting certain DNAs. (They did this by the way.)

I wrote this summary really fast so apologies if it’s not self-explanatory enough. Please feel free to ask questions and leave comments! We’re all here to learn. And to bring accountability and the end of imposed suffering.
This is only the beginning.
The most important thing that I understand about your post (scientific jargon aside) is that you are not stopping to uncover what really took place and make things clear enough that we are all informed … you’re all wonderful souls who we admire so much. Thank you for being you.
So, the very, very few of us that risked everything to warn the masses against taking this garbage, were right! You should write an article on THAT!
I'm still being harassed, stalked, slandered, and threatened because I dared to take a stand from day 1 when no one else seemed to have the courage or intelligence to do so. F--k everyone who didn't speak up and left people like me to carry the burden alone.